
Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
- Сертификация медицинских изделий – что это?
- Основные этапы сертификации лекарств
- Порядок контроля качества лекарств
- Регистрационное удостоверение на лекарства
- Порядок сертификации лекарственных средств в России
- Стоимость получения сертификата
- Какие документы необходимы для получения сертификата на медицинские изделия?
- Медицинские изделия, подлежащие обязательной сертификации
- Какие медицинские изделия подлежат добровольной сертификации?
- Подтверждение соответствия лекарственных средств
- Судебная практика
- Процедура получения сертификата в России
- Документы для сертификации
- Сроки сертификации
- От чего зависит стоимость сертификации ЛС и мед изделий?
- Товары, подлежащие сертификации
- Сертификация лекарств
- Новые правила сертификации лекарственных средств
- Форма обязательной сертификации лекарственных средств
- Процедура государственной регистрации
- Получение регистрационного удостоверения
- Контроль качества препаратов в рамках процедуры государственной регистрации
- Центры сертификации лекарственных средств
- Работа центров сертификации
- Право на выполнение сертификации лекарств
- Процедура сертификации
- К каким производствам применима эта процедура?
- Нормативная база
- Преимущества обладания сертификатом
- Стандарт GMP в международной практике
- Обзор документа
- Правила GMP в России
- Особенности ввода в обращение иммунобиологических лекарственных препаратов (ИЛП)
- Какие документы нужны для получения сертификата на препараты?
- Для чего проводят регистрацию и сертификацию изделий медицинского назначения?
- Где получить сертификат соответствия на медицинские изделия?
- Для чего нужен добровольный сертификат на лекарства?
- Как получить разрешение на ввод в оборот новых лекарственных средств?
Сертификация медицинских изделий – что это?
Лекарственные средства, как и другие медицинские товары, способны повлиять на жизнь и здоровье людей. Поэтому Росздравнадзор тщательно контролирует данные изделия и предусматривает несколько форм их проверки.
Сертификация – обязательная проверка всех медицинских товаров, которые реализуются на территории РФ, произведены они могут быть в любой стране.
Сертификат соответствия (СС) оформляется на бланке установленного образца, содержит следующую информацию:
- Название лекарственного средства или изделия;
- Сведения о заявителе и производителе;
- Нормативы, по которым изготовлен товар;
- Доказательные материалы, подтверждающие безопасность;
- Сведения сертификационного органа;
- Номер документа и дата выдачи.
Сертификат на медицинские изделия и лекарственные средства – это официальный документ, который свидетельствует о соответствии объекта требованиям безопасности. Целью проверки является защита потребителей от некачественных товаров.
Основные этапы сертификации лекарств
Чтобы получить сертификат на указанный вид продукции медицинского назначения, изготовителю или импортеру необходимо пройти следующие стадии:
- обращение в центр для уточнения всех тонкостей проведения процедуры в каждом конкретном случае;
- формирование пакета необходимых документов;
- проведения испытаний в условиях аккредитованной лаборатории и составление протоколов;
- получение готового документа у нашего курьера.
Желаете в оптимальные сроки и на выгодных условиях пройти процедуру сертификации лекарств? Возникли вопросы, касающиеся ввода в обращение лекарств? Обратитесь за помощью в центр Мос РСТ!
Сотрудники компании помогут в оформлении разрешительной документации для законного производства и реализации медицинских товаров.
Практически в каждом регионе РФ работает окружной центр сертификации лекарственных средств, который осуществляет деятельность в области контроля качества, безопасности и действенности таких препаратов в конкретном субъекте Федерации.
Качество лекарственных средств — это вопрос, который находится под пристальным вниманием государства. Федеральная политика в этой области предполагает выработку общих принципов контроля на общегосударственном уровне. Эти принципы дополняются практическими механизмами их реализации в конкретных субъектах. В вопросе сертификации лекарственных средств основную роль играето Росздравнадзоре Он выполняет функции отдела регистрации лекарственных средств, то есть действует в области управления системой регистрации препаратов. Еще одна его задача – это контроль работы территориальных органов по сертификации лекарственных средств, находящихся в регионах Российской Федерации.
В нашей стране предусмотрена обязательная сертификация лекарственных средств, поскольку данная категория товаров может представлять собой повышенный риск возникновения негативных последствий для пациента.
В Российской Федерации в качестве инструмента гарантии безопасности и качества изделий действует система сертификации. Она применяется во всех отраслях экономики и в зависимости от степени потенциальной опасности продукта и других факторов может носить обязательный или добровольный характер. Лекарственные средства относятся в категории товаров, которые могут повлечь за собой повышенный риск появления отрицательных последствий для пациента в виде побочных эффектов и других нежелательных последствий. При этом они могут существенно усугубляться в случае, если препарат не соответствует актуальным требованиям законодательства. Однако для этой группы изделий в нашей стране применяется особый порядок контроля качества.
29 ноября вступает в силу Федеральный закон от 28 ноября 2018 г. N 449-ФЗ “О внесении изменений в отдельные законодательные акты Российской Федерации по вопросу ввода в гражданский оборот лекарственных препаратов для медицинского применения”.
Росаккредитация информирует, что в соответствии со статьей 2 Федерального закона N 449-ФЗ пункт 4 статьи 1 Федерального закона от 27 декабря 2002 г. N 184-ФЗ “О техническом регулировании” после слов “санитарно-эпидемиологических требований,” дополнен словами “требований в сфере обращения лекарственных средств,”. Таким образом, отношения, связанные с разработкой, принятием, применением и исполнением требований в сфере обращения лекарственных средств, выводятся из сферы регулирования Федерального закона N 184-ФЗ.
С 29 ноября 2019 г. также вступают в силу изменения в единый перечень продукции, подлежащей обязательной сертификации, и единый перечень продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии, утвержденные постановлением Правительства Российской Федерации от 1 декабря 2009 г. N 982 (далее – Единые перечни), которые исключают из указанных перечней лекарственные препараты для медицинского применения.
Таким образом, с 29 ноября 2019 г. продукция, исключенная из Единых перечней, обязательной сертификации и декларированию соответствия не подлежит.
Соответствующие настройки начиная с указанной даты будут реализованы в федеральной государственной информационной системе в области аккредитации.
Лекарственным средством называют вещество или смесь веществ натурального или синтетического происхождения, которое выпускается в определенной лекарственной форме: таблетках, инъекциях, мазях и иных. Их используют для лечения различных заболеваний, а также в профилактических целях.
На сегодняшний день сертификация лекарственных средств в России носит добровольный характер. Ранее продукция этого типа входила во второй список, закрепленный Постановлением Правительства № 982, и подлежала обязательному декларированию в рамках системы ГОСТ Р. Но с момента вступления в законную силу Постановления № 489 от 24.04.2019 (а именно, с 29.11.2019) исключена из указанного перечня.
В связи с этим, в настоящее время у предпринимателей нет необходимости оформлять декларацию о соответствии лекарственных средств.
Ввод в обращение товаров этой категории осуществляется после внесения соответствующей информации в АИС Росздравнадзора. Правила прохождения данной процедуры урегулированы ФЗ № 449 от 28.11.2018 и Постановления Правительства № 1510 от 26.11.2019.
Заявитель должен предоставить следующую информацию и документацию на каждую партию или серию продукции:
- сведения о препарате, включающие его наименование, данные, касающиеся производственных площадок и фармацевтических субстанций, используемых в процессе производства;
- документы, подтверждающие качество продукта;
- подтверждение соответствия средства перечню требований, которые установлены при госрегистрации.
Для регулирования обращения медицинских препаратов была создана система мониторинга данной продукции. Доступ к базе данных для производителей этой категории товаров является бесплатным. Изготовление и реализация лекарств без нанесения на них средств идентификации (с нарушением правил нанесения), а также невнесение данных в систему мониторинга служат основанием для применения мер административной ответственности.
Что касается ввозимых на территорию РФ лекарственных препаратов, они должны в обязательном порядке сопровождаться разрешительной документацией иностранного производителя.
Более подробно о том, какие разрешения необходимы для законного выпуска, импорта и реализации лекарственных препаратов, вы узнаете, обратившись к специалистам центра сертификации.
Процедуру сертификации лекарств изготовитель или импортер может пройти исключительно по собственной инициативе в добровольном порядке.
Порядок контроля качества лекарств
До недавнего времени в Российской Федерации действовало обязательное правило о сертификации лекарственных препаратов. Они подлежали процедуре контроля на общих основаниях: то есть проверка качества проводилась в отношении тех позиций, которые были упомянуты в постановлении Правительства № 982. Такое условие распространялось на следующие категории препаратов:
- медикаменты и продукты химического и фармацевтического профиля;
- витамины и аминокислоты;
- ферменты и коферменты;
- эндокринные препараты;
- ряд категорий лекарств и фармакологических продуктов, предназначенных для использования в ветеринарии;
- биологическое сырье, питательные основы и среды;
- бактериофаги и аллергены;
- другие виды продуктов.
Чаще всего процедура сертификации перечисленных позиций выполнялась в форме декларирования. Выполнение проверок этих и других препаратов на соответствие установленным нормативам проводилось на оснвоании постановления Госстандарта от 24 мая 2002 года N 36. При этом из правила о выполнении обязательной сертификации существовал ряд исключений. Их список, приведенный в письме Госстандарта от 15 января 2003 года N ИК-110-25/110, включал:
- лекарства, не имеющие индивидуальной упаковки и не предназначенные для продажи в розницу (для этой группы препаратов использовалось особое наименование – «ин балк»);
- фармацевтические субстанции для использования в рамках технологического цикла по производству готовых лекарств;
- вакцины, сыворотки и иммунобиологические средства, не входящие в список препаратов, требующих обязательной сертификации.
Регистрационное удостоверение на лекарства
Регистрационное удостоверение является официальным документом, который подтверждает, что препараты соответствуют требованиям в отношении их безопасности и эффективности. РУ на лекарственные средства выдается по итогам успешного прохождения комплекса исследований, (токсикологических, клинических, технических). Указанный документ действует бессрочно.
Процедура регистрации этой разновидности медицинских товаров осуществляется органами Росздравнадзора, на ее проведение может потребоваться от 1 года до 1,5 лет.
Для оформления регистрационного удостоверения предпринимателю потребуется:
- подготовить комплект необходимой документации;
- получить направление в Росздравнадзоре на клинические испытания продукции;
- пройти комплекс исследований и получить протоколы по их результатам;
- предоставить подтверждение качества и безопасности медикаментов в Росздравнадзор;
- получить оформленное удостоверение.
Если у вас возникли трудности в процессе подготовки документации или получении направления на тестирование лекарств, воспользуйтесь услугами центра сертификации.
Порядок сертификации лекарственных средств в России
Лекарственные средства имеют стандартный порядок сертификации, он включает в себя:
- Выбор сертифицирующего органа – это может быть сторонняя организация с соответствующей аккредитацией;
- Выбор схемы оценки;
- Отбор образцов для тестирования;
- Идентификация изделий;
- Испытания в аккредитованной лаборатории с составлением протокола полученных показателей;
- Оценка производства (если предусмотрено схемой);
- Анализ полученных сведений и материалов, принятие решения;
- Выдача сертификата или мотивированного отказа;
- Инспекционные контроль (если предусмотрено).
Предусмотрено несколько схем сертификации:
- 7с – на партию;
- 3 и 3а – на серийное производство.
Срок действия документа ограничен – до 3-х лет, после чего требуется повторная процедура прохождения оценки изделий.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
Какие документы необходимы для получения сертификата на медицинские изделия?
Перечень необходимых документов состоит из:
- Заявления установленного образца;
- Регистрационное удостоверение;
- Копию учредительных и организационных документов;
- Копия устава;
- Протоколы проведенных испытаний;
- Образцы изделий;
- Технические условия;
- Разрешение на применение товара в лечебной практике;
- Доверенность от компании-изготовителя на предоставление своих интересов на территории РФ (для импортных товаров);
- Подтверждающие безопасность изделия материалы;
- Другие по запросу.
Все копии должны быть заверенными, пакет документов сшит.
Медицинские изделия, подлежащие обязательной сертификации
Практически вся медицинская продукция подлежит либо обязательной сертификации, либо добровольному декларированию. Перечни изделий и порядок проведения определены Постановлением Правительства №982 и Правилами №36. Нужно оформлять СС на изделия:
- Лекарственные средства, выпускаемые на территории РФ;
- Ввозимые на территорию РФ.
Отсутствие документа является административным нарушением, влечет штрафы и изъятие продукции.
Какие медицинские изделия подлежат добровольной сертификации?
Лекарственные изделия, на которые можно оформить добровольный СС:
- Средства без индивидуальной упаковки, созданные не для прямой реализации;
- Субстанции для производства лекарств;
- Вакцины, сыворотки, иммунобиологические средства.
Добровольный и обязательный документы идентичны, одинаковы порядки их оформления и регистрации. В самом бланке СС есть указание, обязательна ли форма оформления.
Подтверждение соответствия лекарственных средств
Подборка наиболее важных документов по запросу Подтверждение соответствия лекарственных средств (нормативно–правовые акты, формы, статьи, консультации экспертов и многое другое).
Судебная практика
Подборка судебных решений за 2020 год: Статья 4 “Основные понятия, используемые в настоящем Федеральном законе” Федерального закона “Об обращении лекарственных средств””Государственная регистрация лекарственного средства является разрешительной процедурой его обращения на территории России и не предоставляет заявителю дополнительных прав на зарегистрированное лекарственное средство. Факт государственной регистрации лекарственного препарата свидетельствует о том, что зарегистрированный лекарственный препарат, производимый указанным в регистрационном удостоверении производителем, может легально обращаться на территории Российской Федерации. Процедура государственной регистрации лекарственных препаратов, предусмотренная Законом N 61-ФЗ, направлена на подтверждение соответствия выпускаемого лекарственного препарата установленным нормам и правилам и не направлена на удостоверение исключительных прав лица, выпускающего соответствующую продукцию. Регистрационное удостоверение на лекарственные препараты представляет собой документ, который официально подтверждает факт соответствия лекарства тем техническим характеристикам, которые заявлены производителем. Этот документ гарантирует качество медицинского препарата и его безопасность в использовании и употреблении.”
Федеральный закон от 27.12.2002 N 184-ФЗ(ред. от 02.07.2021)”О техническом регулировании”(с изм. и доп., вступ. в силу с 23.12.2021)11. До перехода к производству лекарственных средств по правилам организации производства и контроля качества лекарственных средств в соответствии со статьей 45 Федерального закона от 12 апреля 2010 года N 61-ФЗ “Об обращении лекарственных средств” обязательное подтверждение соответствия лекарственных средств осуществляется в соответствии с нормативными правовыми актами Российской Федерации и нормативными документами федеральных органов исполнительной власти, указанными в пунктах 1 и 2 настоящей статьи и применяемыми в части, не урегулированной указанным Федеральным законом.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
От чего зависит стоимость сертификации ЛС и мед изделий?
Стоимость процедуры рассчитывается индивидуально. Факторы, от которых зависит цена:
- Вид продукции;
- Класс опасности товаров;
- Размер предприятия;
- Схема сертификации;
- Наличие документации.
Для консультации по поводу цены и срока оформления документа можно обратиться к экспертам сертифицирующего центра. Каждый центр предлагает комплексную помощь для получения СС.
Товары, подлежащие сертификации
Общий список товаров, в отношении которых в нашей стране применяется условие об обязательной сертификации, приведен в постановлении Правительства № 982. Рассматриваемый нормативный документ периодически пересматривается, чтобы обеспечить соответствие его содержания реальной ситуации на рынке. Чаще всего это делается не реже одного раза в год, а иногда и по несколько раз за этот период. Например, это возможно в случае, если участились ситуации отравлений или проявления иных признаков недоброкачественности какого-то продукта, он может быть внесен в список позиций, требующих обязательной сертификации.
Иногда происходит и наоборот: конкретные товары исключаются из списка, содержащегося в постановлении. Обычно это случается, если в этой части рынка сложилась достаточно благоприятная ситуация с минимумом претензий к этому типу продукта. Другое распространенное основание – введение нового механизма контроля для конкретного вида изделий.
Сертификация лекарств
Совсем недавно значительная часть лекарственных препаратов также была отражена в постановлении № 982, то есть требовала обязательной сертификации. Такое условие распространялось на такие препараты как витамины, ферменты, эндокринные лексредства и ряд других. При этом в отношении большей части из них контроль качества должен был выполняться в форме декларирования. Это особый механизм контроля, который предполагает, что вся ответственность за характеристики товара возлагается на производителя. Он формирует декларацию соответствия, в которой указывает, что продукт отвечает государственным нормативам по безопасности и составу товара, а уполномоченная сертификационная организация проверяет препарат и регистрирует декларацию.
При этом для отдельных категорий таких продуктов условие об обязательной проверке качества было отменено письмом Госстандарта от 15 января 2003 года N ИК-110-25/110. Например, в России разрешалось применять несертифицированные лекарства «ин балк», то есть без потребительской упаковки, если они не предполагаются для продажи в розницу, а также некоторые другие типы лекарственных средств. Однако недавно этот порядок был серьезно изменен.
Новые правила сертификации лекарственных средств
В конце прошлого года в силу вступило постановление Правительства от 26 ноября 2019 года N 1510. Этот документ формально отменил требование об обязательной сертификации лексредств. Однако профессиональные участники рынка, конечно, понимали: эта категория товаров не может поступать в аптечные организации без серьезного контроля. И механизмы такого контроля в новом порядке прописаны четко
На самом деле обновленные правила систематизировали и упорядочили в некоторой степени фрагментарные инструменты контроля, которые применялись до этого. Согласно федеральному закону № 61-ФЗ в России для того, чтобы лекарство попало на рынок, необходимо пройти процедуру государственной регистрации. В ходе нее заявитель оформляет и направляет в контролирующее ведомство множество документов о своем продукте и многократно доказывает, что он отвечает актуальным нормативам по качеству, безопасности и действенности. Однако до недавнего времени производители должны были проходить еще и процедуру сертификации, которая фактически ориентирована на те же цели. А дополнительные контролирующие меры – это, конечно, трата финансовых ресурсов и времени, в течение которого препарат мог бы уже лежать на прилавках и приносить прибыль производителю.
Форма обязательной сертификации лекарственных средств
Процедура обязательной сертификации лекарственных средств в России выполняется в форме регистрации медицинских препаратов. Она проводится Росздравнадзором по инициативе заявителя, в качестве которого выступают как сами производители таких продуктов, так и организации, занимающиеся ее распространением. При этом регистрация становится обязательной для большей части лекарств, созданных для широкого обращения на рынке фармацевтических товаров.
Процедура государственной регистрации
Сейчас законодатели пришли к выводу, что тех проверочных мер, которые входят в процедуру госрегистрации, будет достаточно для организации адекватного контроля за этим сегментом рынка. Она по-прежнему будет выполняться с соблюдением регламента, прописанного в приказе Минздрава от 21 сентября 2016 года N 725н. В роли государственного ведомства, которое оказывает эту услугу, выступает Росздравнадзор.
Сама процедура регистрации включает два базовых этапа:
- подготовительный. На этом шаге всю работу делает заявитель. Правда, он может обратиться в надзорное ведомство за консультацией, если она ему потребуется – например, для организации клинических исследований. В течение подготовительной фазы заявитель проходит проверку Минпромторга на соответствие правилам надлежащей производственной практики, составляет регистрационное досье в формате общетехнического документа (ОТД) и проводит клинические исследования препарата. Выполнение каждой процедуры должно быть подтверждено документально. Сформированный пакет документов представляется в Росздравнадзор для инициирования процедуры госрегистрации;
- основной. На этом шаге в действие вступают две основные государственные структуры. В роли координатора процесса функционирует Росздравнадзор ,который проверяет документы, полученные от заявителя, и составляет задание на выполнение необходимых экспертиз. Сами проверки и исследования в рамках этих экспертиз проводит отдельная уполномоченная организация – ФГБУ «НЦЭСМП». Она работает только по заданию Росздравнадзора, то есть прямые контакты с заявителями для нее запрещены. В рамках этого этапа проводится два типа экспертиз: проверка безопасности и качества препарата, а также проверка по шкале «риск-польза». В процессе этого исследования выполняется сопоставление предполагаемого риска, обусловленного использованием препарата, и ожидаемой пользы от его применения в конкретной врачебной ситуации.
Получение регистрационного удостоверения
Изделие, которое прошло все требуемые проверки и подтвердило свое соответствие актуальным нормативам по качеству и безопасности этой категории товаров, получает регистрационное удостоверения. Оно становится документальным подтверждением того, что конкретное лекарство зарегистрировано и имеет прав продаваться на рынке. Информация об этом также отражается в едином государственном реестре. Он опубликован в открытом доступе на официальном портале Министерства здравоохранения.
Описанный порядок действовал до 2019 года. Однако затем было принято постановление Правительства от 26 ноября 2019 года N 1510, которое установило новые правила ввода лекарств, предназначенных для использования в медицине, в гражданский оборот. Одним из основных нововведений, зафиксированных в этом постановлении, стала отмена процедуры сертификации лекарственных средств. Однако это не предполагает, что государственный контроль за качеством и безопасностью этой жизненно важной категории полностью ликвидирован.
Новое постановление систематизировало алгоритм реализации установленных обязательных действий, которые имеют общую цель – выполнение контроля за качеством лекарств для гарантии их безопасности. Теперь все процедуры в рамках контроля, надзора и учета в этой области переданы Росздравнадзору.
Контроль качества препаратов в рамках процедуры государственной регистрации
Согласно правилам, зафиксированным в недавно принятом постановлении № 1510 проверка лекарств на соответствие нормативам вошла в состав процедуры государственной регистрации лекарств. Она предусматривается положениями федерального закона № 61, который устанавливает порядок обращения лекпрепаратов в России. В рамках этой процедуры реализуется многоуровневый подход к анализу характеристик конкретного препарата, включающий следующие шаги:
- оформление сертификата GMP, подтверждающего соответствие производителя требованиям надлежащей производственной практики. Проверка по этим параметрам выполняется инспекторами Минпромторга, которые также оформляют и выдают сертификат;
- выполнение клинических исследований препарата заявителем на этапе подготовки к процедуре регистрации. Результаты исследований оформляются в виде отчета установленного образца;
- составление регистрационного досье в формате общетехнического документа (ОТД), включающего все основные сведения о препарате;
- организацию экспертизы качества лекарства, в рамках которой также анализируется действенность механизмов контроля этого параметра, предложенных производителем. Эта процедура сегодня выполняется единственной уполномоченной организацией – ФГБУ «НЦЭСМП»;
- организация экспертизы по шкале «риск-польза», в рамках которой оценивается потенциальный риск, связанный с использованием препарата, и предполагаемая польза в рамках конкретной медицинской цели. Этот тип экспертизы также проводится ФГБУ «НЦЭСМП».
В отдельныз ситуациях постановление требует выполнения дополнительных контрольных процедур. Например, на первые три партии или серии препарата, который впервые ввозится в Россию или впервые изготавливается на ее территории, нужно будет оформить еще и специальный протокол, который подтверждает соответствие этого средства актуальным требованиям.
Итак, невзирая на формальную отмену сертификации лексредств, правила контроля их качества, безопасности и действенности остаются строгими. Это нужно, чтобы исключить попадание опасных, неэффективных или фальсифицированных продуктов на рынок.
Центры сертификации лекарственных средств
Сейчас практически в каждом регионе РФ функционирует окружной центр сертификации лекарственных средств, который работает в области контроля качества, безопасности и действенности таких препаратов в конкретном субъекте Федерации. Основной целью их деятельности становится анализ обращающихся на рассматриваемой территории медпрепаратов.
Работа центров сертификации
Основными направлениями работы стандартного территориального органа регистрации и сертификации лекарственных средств становятся:
- выполнение исследований и испытаний лекарственных средств в областях, отвечающих параметрам аккредитации конкретной организации;
- сбор и анализ сведений о качестве и иных параметрах препаратов, ввозимых на территорию региона из других субъектов Российской Федерации или из-за рубежа;
- обеспечение методической, консультационной и организационной поддержки организациям, занимающимся выпуском или распространением лекарственных средств на территории региона.
Такие центры могут выполнять необходимые виды работ на стадии подготовки препарата к процедуре государственной регистрации. Кроме этого, важное направление их работы – выполнение контроля качества лекарств. Он может проводиться:
- по инициативе покупателя, например, в случае появлениия у последнего сомнений в качестве средства или его подлинности. При этом в такой ситуации покупатель может обратиться не только в аккредитованный сертификационный центр, но и прямо в территориальное подразделение Росздравнадзора по месту своего пребывания;
- по инициативе производителя товара, который собирается организовать проверку своего препарата в рамках одного из действующих стандартов или систем добровольной сертификации. Чаще всего производители идут на такой шаг в целях организации рекламной поддержки своего товара или при выходе на зарубежные рынки.
Процедура сертификации включает анализ качественных параметров препарата, степень его безопасности и действенности с использованием специализированного оборудования в условиях лаборатории. Проверка лексредства осуществляется подготовленными экспертами, имеющими профильное образование и опыт работы в этой области. Для организации контролирующих мероприятий используется несколько единиц образцов лекпрепарата, чтобы обеспечить надежность выполненных измерений и исследований.
Право на выполнение сертификации лекарств
Основное условие, которое дает сертификационному органу право выполнять экспертизу лекарственных средств, – это наличие соответствующего пункт в его области аккредитации. Чтобы получить аккредитацию с требуемыми параметрами, компании нужно обратиться в Росаккредитацию, предоставив документы, подтверждающие наличие у нее необходимых компетенций и соответствие иным требованиям.
Информацию об аккредитованных сертификационных органах и областях их аккредитации можно найти на портале ведомства в открытом доступе. Это сделано, чтобы потенциальные заказчики и другие заинтересованные лица могли проконтролировать, что исполнитель, которого они выбрали для проведения экспертизы лекпрепаратов, в самом имеет нужные полномочия. Кроме того, некоторые данные о таких организациях в справочном режиме опубликованы и на сайте Росздравнадзора: например, здесь можно обнаружить контактные сведения аккредитованных компаний, работающих в Москве, Московской области и Санкт-Петербурге.
Процедура сертификации
Общие правила сертификации в Российской Федерации определены положениями федерального закона № 184-ФЗ, посвященного вопросам технического регулирования. Этот нормативный документ устанавливает, что в отношении ряда товаров в нашей стране применяется условие об обязательной сертификации. Чаще всего такое условие распространяется на следующие категории товаров:
- которые могут стать потенциально опасными для покупателя в случае нарушения правил их производства или иных нормативов;
- производство которых является сложным и многоуровневым, что обусловливает необходимость тщательного контроля готовой продукции на предмет соблюдения всех актуальных нормативов.
Сама процедура сертификации выглядит как проверка образцов продукта на соответствие установленным нормативам по качеству. Эти требования зафиксированы в особых нормативных документах, разработанных для конкретной категории товаров. В России в этом качестве часто применяются национальные, межгосударственные или международные стандарты ГОСТ Р.
Проводить такую проверку имеют право только проверенные уполномоченные организации, которые прошли процедуру аккредитации, подтвердив этим уровень своей компетентности. При этом область аккредитации такой организации должна включать выполнение контроля именно для той категории товаров, которые она планирует исследовать. Проверка проводится с использованием уникального лабораторного оборудования, которое позволяет выполнить все испытания, предусмотренные нормативами для этой категории товаров.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
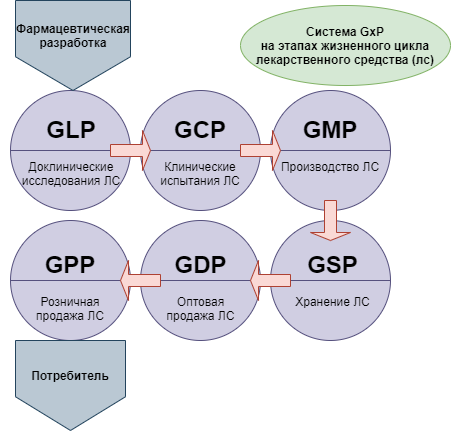
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Обзор документа
С 29 ноября 2019 г. действуют законодательные поправки по вопросу ввода в гражданский оборот лекарственных препаратов для медицинского применения.
Из сферы действия Закона о техническом регулировании исключены разработка, принятие, применение и исполнение требований по обращению лекарственных средств.
Кроме того, лекарственные препараты исключены из перечней сертифицируемой и декларируемой продукции.
Для просмотра актуального текста документа и получения полной информации о вступлении в силу, изменениях и порядке применения документа, воспользуйтесь поиском в Интернет-версии системы ГАРАНТ:
Правила GMP в России
Особенности ввода в обращение иммунобиологических лекарственных препаратов (ИЛП)
После получения доступа к АИС изготовитель или импортер должен заполнить заявление, указав детальную информацию об ИЛП, к нему прикрепляют отсканированные в формате pdf:
- заключение ФГБУ «НЦЭСМП» Минздрава России (или ФГБУ «ИМЦЭУАОСМП» Росздравнадзора);
- протоколы проведенных испытаний образцов продукции.
Затем заявление рассматривается Росздравнадзором, информацию о предоставлении разрешения или об отказе можно найти на его официальном сайте в соответствующем разделе «Реестр разрешений».
Какие документы нужны для получения сертификата на препараты?
Сертификация лекарственных средств проводится при условии предоставления субъектом предпринимательской деятельности следующего комплекта документации:
- копий учредительных и регистрационных документов;
- технической документации – ГОСТа, ТУ;
- описания продукта – включает наименование, группу, код, состав и иные характеристики;
- инструкцию по использованию;
- договор на аренду производственных площадей или свидетельство о праве собственности;
- контракт о поставке медпрепаратов с инвойсами, спецификациями и другими приложениями на импортируемые из других стран средства;
- протоколы испытаний – при наличии;
- копию регистрационного удостоверения;
- иные документы – сертификаты ИСО, экспертные заключения и другие.
Для чего проводят регистрацию и сертификацию изделий медицинского назначения?
Основной целью оформления разрешительного документа является выполнения требований законодательства страны. Его наличие позволит легко проходить проверки, которые обязательны для медицинских товаров. Отсутствие СС чревато штрафами и изъятием партии. Среди других причин:
- Повышение лояльности со стороны потребителей;
- Формирование положительного имиджа среди компаний-конкурентов;
- Укрепление своих позиций;
- Возможность подачи заявки на тендерах федерального масштаба;
- Увеличение рынков сбыта;
- Увеличение продаж.
Где получить сертификат соответствия на медицинские изделия?
Заниматься оформлением сертификатов по ГОСТу может не любая организация, а только имеющая соответствующую аккредитацию. Процедура подразумевает участие трех сторон: заявителя (чаще всего это производитель, но может быть и поставщик), сертифицирующего органа и испытательной лаборатории. Чтобы не ошибиться с выбором исполнителя, стоит обратить внимание на:
- Опыт работы организации;
- Спектр медицинских товаров, с которыми проводятся процедуры;
- Наличие собственной испытательной лаборатории;
- Гибкое ценообразование;
- Разумные сроки выполнения оценки (слишком быстрые сроки могут быть сомнительны).
Для чего нужен добровольный сертификат на лекарства?
Подтверждение соответствия препаратов проводится в зарегистрированной системе добровольной сертификации. По результатам данной процедуры заявителю выдается добровольный сертификат (декларация на лекарственные средства не оформляется). Срок действия указанного документа не может превышать 3 лет.
Несмотря на то, что наличие сертификата соответствия на медикаменты не является обязательным требованием действующего законодательства, многие предприниматели заинтересованы в его получении. Наличие данного документа:
- способствует росту конкурентоспособности;
- повышает доверие широкого круга потребителей;
- увеличивает шанс выигрыша при участии в государственных закупках.
Сертификация предусматривает обязательное исследование образцов товара. В лаборатории проводят экспертизы и составляют протокол испытаний. На основании данных протокола оформляется сертификат соответствия.
Как получить разрешение на ввод в оборот новых лекарственных средств?
Все лекарства, на которые регистрационное удостоверение было выдано после 28.11.2019, являются впервые производимыми или впервые ввозимыми. Для того чтобы получить разрешение на ввод в обращение товаров этой категории, необходимо:
До внесения соответствующих протоколов на препараты, на которые РУ было получено после 28.11.2019, сведения о партии или серии товаров будут находиться в статусе ожидания. Опубликуются они только после того, как заявитель прикрепит соответствующие протоколы.