
Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
Процедура регистрации лекарственных средств является обязательной для легитимного обращения медицинских препаратов на территории Российской Федерации.
Согласно федеральному закону № 61-ФЗ процедура регистрации лекарственных средств обязательна для легитимного обращения медицинских препаратов на территории Российской Федерации. При этом одним из ключевых ее этапов становится выполнение экспертизы лекарств. Эта процедура достаточно требует серьезных вложений времени и финансовых средств, поскольку она включает несколько сложных и продолжительных исследований.
В нашей стране предусмотрена обязательная сертификация лекарственных средств, поскольку данная категория товаров может представлять собой повышенный риск возникновения негативных последствий для пациента.
В Российской Федерации в качестве инструмента гарантии безопасности и качества изделий действует система сертификации. Она применяется во всех отраслях экономики и в зависимости от степени потенциальной опасности продукта и других факторов может носить обязательный или добровольный характер. Лекарственные средства относятся в категории товаров, которые могут повлечь за собой повышенный риск появления отрицательных последствий для пациента в виде побочных эффектов и других нежелательных последствий. При этом они могут существенно усугубляться в случае, если препарат не соответствует актуальным требованиям законодательства. Однако для этой группы изделий в нашей стране применяется особый порядок контроля качества.
29 ноября вступает в силу Федеральный закон от 28 ноября 2018 г. N 449-ФЗ “О внесении изменений в отдельные законодательные акты Российской Федерации по вопросу ввода в гражданский оборот лекарственных препаратов для медицинского применения”.
Росаккредитация информирует, что в соответствии со статьей 2 Федерального закона N 449-ФЗ пункт 4 статьи 1 Федерального закона от 27 декабря 2002 г. N 184-ФЗ “О техническом регулировании” после слов “санитарно-эпидемиологических требований,” дополнен словами “требований в сфере обращения лекарственных средств,”. Таким образом, отношения, связанные с разработкой, принятием, применением и исполнением требований в сфере обращения лекарственных средств, выводятся из сферы регулирования Федерального закона N 184-ФЗ.
С 29 ноября 2019 г. также вступают в силу изменения в единый перечень продукции, подлежащей обязательной сертификации, и единый перечень продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии, утвержденные постановлением Правительства Российской Федерации от 1 декабря 2009 г. N 982 (далее – Единые перечни), которые исключают из указанных перечней лекарственные препараты для медицинского применения.
Таким образом, с 29 ноября 2019 г. продукция, исключенная из Единых перечней, обязательной сертификации и декларированию соответствия не подлежит.
Соответствующие настройки начиная с указанной даты будут реализованы в федеральной государственной информационной системе в области аккредитации.
- Процедура сертификации
- Стандарт GMP в международной практике
- Подготовка к выполнению экспертизы
- Правила выполнения экспертизы
- Организация исследований
- Ускоренное выполнение экспертизы
- Правила составления и применения экспертного заключения
- Повторное выполнение экспертизы
- Применение результатов экспертизы
- Процедура получения сертификата в России
- Документы для сертификации
- Сроки сертификации
- Стоимость получения сертификата
- К каким производствам применима эта процедура?
- Нормативная база
- Преимущества обладания сертификатом
- Правила GMP в России
- Товары, подлежащие сертификации
- Сертификация лекарств
- Новые правила сертификации лекарственных средств
- Форма обязательной сертификации лекарственных средств
- Процедура государственной регистрации
- Получение регистрационного удостоверения
- Хранение сертификатов
- Сертификация лс.
- Значение экспертизы лекарств
- Обзор документа
Процедура сертификации
Общие правила сертификации в Российской Федерации определены положениями федерального закона № 184-ФЗ, посвященного вопросам технического регулирования. Этот нормативный документ устанавливает, что в отношении ряда товаров в нашей стране применяется условие об обязательной сертификации. Чаще всего такое условие распространяется на следующие категории товаров:
- которые могут стать потенциально опасными для покупателя в случае нарушения правил их производства или иных нормативов;
- производство которых является сложным и многоуровневым, что обусловливает необходимость тщательного контроля готовой продукции на предмет соблюдения всех актуальных нормативов.
Сама процедура сертификации выглядит как проверка образцов продукта на соответствие установленным нормативам по качеству. Эти требования зафиксированы в особых нормативных документах, разработанных для конкретной категории товаров. В России в этом качестве часто применяются национальные, межгосударственные или международные стандарты ГОСТ Р.
Проводить такую проверку имеют право только проверенные уполномоченные организации, которые прошли процедуру аккредитации, подтвердив этим уровень своей компетентности. При этом область аккредитации такой организации должна включать выполнение контроля именно для той категории товаров, которые она планирует исследовать. Проверка проводится с использованием уникального лабораторного оборудования, которое позволяет выполнить все испытания, предусмотренные нормативами для этой категории товаров.
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Подготовка к выполнению экспертизы
Выполнение экспертизы лекарства – это самый ответственный этап процедуры госрегистрации, до которого доходят далеко не все заявители. Для отсеивания заявок, которые не соответствуют требованиям законодательства, федеральным законом № 61-ФЗ предусмотрена предварительная подача заявителем пакета документации на препарат для рассмотрения экспертами Минздрава. Этот пакет включает следующие документы:
- сертификат GMP, который играет роль документального подтверждения того, что производитель, подающий заявку на регистрацию своего продукта, соответствует актуальным правилам надлежащей производственной практики. Такое условие введено в законодательство относительно недавно – с 2016 года. Проверку на соответствие рассматриваемым требованиям проводят инспекторы Минпромторга, и они же оформляют нужный сертификат;
- отчет о выполненных клинических исследованиях. Этот документ входит в состав пакета, передаваемого на регистрацию, если выполнение клинических исследований необходимо для конкретного препарата. Из этого правила есть ряд исключений – они перечислены в статье 18 федерального закона № 61-ФЗ;
- регистрационное досье, сформированное согласно принципам, прописанными в приказе Минздрава от 12 июля 2017 года N 409н. Требования этого нормативного акта предписывают, чтобы оно оформлялось в форме общетехнического документа (ОТД).
Подготовленный пакет вместе с отобранными образцами продукта передается в Минздрав. Там в течение десяти рабочих дней уполномоченные эксперты проводят анализ сформированной заявки на предмет корректности составления документов и полноты комплекта. Если с этим все в порядке, назначается процедура экспертизы лексредства.

Правила выполнения экспертизы
Необходимые этапы исследования лекарственных препаратов, претендующих на регистрацию на территории РФ , зафиксированы в ст. 14 ФЗ-61. В их число входят следующие стадии:
- анализ методики оценки уровня качества конкретного лексредства и использование предложенной методики для реализации такого контроля;
- анализ соотношения предполагаемой пользы от применения лекарства к возможным отрицательным следствиям его использования.
Для препаратов, которые представлены на регистрацию в качестве орфанных, то есть предназначенных для лечения редких типов заболеваний, также выполняется анализ документации, которая подтверждает возможности их использования для этих медицинских целей.
Организация исследований
Выполнять требуемые исследования имеет право только одно уполномоченное предприятие, подконтрольное Министерству здравоохранения. Это ФГБУ «НЦЭСМП». При этом оно вправе выполнять аналитические процедуры только по заданию Минздрава: прямое взаимодействие с заявителями в рамках договорных отношений не разрешается. Такое взаимодействие ограничено даже в тех ситуациях, когда уполномоченная организация сталкивается с нехваткой материалов или информации для организации экспертизы. В этой ситуации руководитель предприятия обращается с таким запросом в Минздрав, который уже запрашивает у заявителя недостающие сведения. Такой механизм выработан для гарантии объективности проводимых исследований и отсутствия возможности воплощения коррупционных схем в ходе выполнения экспертизы.
Это – одно из основных условий, применяемых при выполнении экспертиз. Этот и другие принципы зафиксированы в основном нормативном документе, который регулирует порядок их осуществления: это приказ Минздрава от 24.08.2017 № 558н. В этом приказе зафиксировано, что эксперт, выполняющий анализ препарата и документации на него, не может находиться в какой-то зависимости от любого лица, которое заинтересовано в его результатах – будь то сотрудник Министерства здравоохранения, изготовитель препарата или другое лицо. При этом за составление ложного, фальсифицированного или преднамеренно неполного экспертного заключения эксперт привлекается к ответственности согласно действующему законодательству.
Ускоренное выполнение экспертизы
По действующему правилу общая длительность процедуры регистрации лекпрепарата в России составляет 160 рабочих дней. В этот период включается и организация всех необходимых экспертиз. Однако в отдельных ситуациях ход осуществления аналитических процедур может быть еще ускорен: проверка документов, составляющих регистрационное досье, также выполняется в течение десяти рабочих дней, но на выполнение обеих видов экспертиз отводится только 60 рабочих дней. В обычном случае на эту процедуру согласно пункту 27 приказа № 558н отводится 110 рабочих дней. Ускоренный порядок применяется для следующих категорий лекарств:
- орфанные препараты, использующиеся для лечения редких типов заболеваний;
- первые три дженерика оригинального лексредства, регистрируемые в России;
- лекарства, предназначенные только для лечения детей, не достигших 18 лет.
Правила составления и применения экспертного заключения
Выводы экспертов относительно качества направленного на регистрацию препарата, а также соотношения «риск-польза» оформляются в виде экспертного заключения. Оно прописывается по форме, приведенной в приложениях к приказу № 558н. В приложениях к этому нормативному документу также содержатся формы дополнительных документов – например, для оценки степени взаимозаменяемости препарата.
Основным результатом экспертизы, который приводится в заключении, становится оценка степени безопасности, действенности и качества препарата. По результатам выполненной аналитической работы эксперт может прийти к выводу о том, что определить значения этих параметров невозможно. Однако в любой ситуации уполномоченное учреждение не принимает самостоятельных решений относительно исхода процедуры регистрации. Оно только передает сформированное заключение в Минздрав, который уже проводит анализ выводов экспертов и выносит положительное или отрицательное решение по поводу госрегистрации.
Повторное выполнение экспертизы
Чаще всего отказ в государственной регистрации по результатам проведенной экспертизы окончателен. Однако вместе с тем ст. 25 ФЗ-61 предусматривает отдельные ситуации, в которых производитель имеет право обратиться за назначением повторной экспертизы с отменой результатов ранее осуществленного исследования. Такое право у него возникает в тех случаях, когда результаты первой процедуры оказались недостоверными не по его вине. Речь идет о следующих ситуациях:
- необоснованность, неполнота или противоречивость заключения;
- фальсификация результатов экспертизы;
Во всех этих случаях производитель лекарственного средства имеет право подать заявление на назначение повторного исследования. В случае его утверждения этого заявления повторная процедура должна быть осуществлена в срок, не превышающий 30 рабочих дней. При этом, поскольку недостоверные результаты были получены не по вине заявители, он освобождается от обязанности по повторной оплате проводимых действий.
Применение результатов экспертизы
После того, как Минздрав получил результаты экспертизы, он обязан в срок, составляющий не более 10 рабочих дней, осуществить обработку и анализ заключения и произвести следующие операции:
- опубликовать на портале министерства переданные заключения и оценить их на предмет соответствия первоначально выданному заданию;
- вынести решение по поводу регистрации лексредства или отказе в его государственной регистрации;
- в случае, если по вопросу регистрации вынесено положительное решение – внести информацию об этом факте в государственный реестр лексредств.
Если Министерство отказало в регистрации по причине содержащейся в экспертном заключении информации о недостаточной безопасности, действенности или уровне качества препарата, изготовитель вправе подать повторное заявление, внеся необходимые изменения в состав средства.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
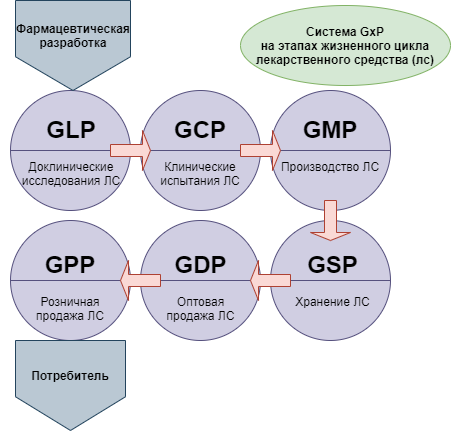
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Правила GMP в России
Товары, подлежащие сертификации
Общий список товаров, в отношении которых в нашей стране применяется условие об обязательной сертификации, приведен в постановлении Правительства № 982. Рассматриваемый нормативный документ периодически пересматривается, чтобы обеспечить соответствие его содержания реальной ситуации на рынке. Чаще всего это делается не реже одного раза в год, а иногда и по несколько раз за этот период. Например, это возможно в случае, если участились ситуации отравлений или проявления иных признаков недоброкачественности какого-то продукта, он может быть внесен в список позиций, требующих обязательной сертификации.
Иногда происходит и наоборот: конкретные товары исключаются из списка, содержащегося в постановлении. Обычно это случается, если в этой части рынка сложилась достаточно благоприятная ситуация с минимумом претензий к этому типу продукта. Другое распространенное основание – введение нового механизма контроля для конкретного вида изделий.
Сертификация лекарств
Совсем недавно значительная часть лекарственных препаратов также была отражена в постановлении № 982, то есть требовала обязательной сертификации. Такое условие распространялось на такие препараты как витамины, ферменты, эндокринные лексредства и ряд других. При этом в отношении большей части из них контроль качества должен был выполняться в форме декларирования. Это особый механизм контроля, который предполагает, что вся ответственность за характеристики товара возлагается на производителя. Он формирует декларацию соответствия, в которой указывает, что продукт отвечает государственным нормативам по безопасности и составу товара, а уполномоченная сертификационная организация проверяет препарат и регистрирует декларацию.
При этом для отдельных категорий таких продуктов условие об обязательной проверке качества было отменено письмом Госстандарта от 15 января 2003 года N ИК-110-25/110. Например, в России разрешалось применять несертифицированные лекарства «ин балк», то есть без потребительской упаковки, если они не предполагаются для продажи в розницу, а также некоторые другие типы лекарственных средств. Однако недавно этот порядок был серьезно изменен.
Новые правила сертификации лекарственных средств
В конце прошлого года в силу вступило постановление Правительства от 26 ноября 2019 года N 1510. Этот документ формально отменил требование об обязательной сертификации лексредств. Однако профессиональные участники рынка, конечно, понимали: эта категория товаров не может поступать в аптечные организации без серьезного контроля. И механизмы такого контроля в новом порядке прописаны четко
На самом деле обновленные правила систематизировали и упорядочили в некоторой степени фрагментарные инструменты контроля, которые применялись до этого. Согласно федеральному закону № 61-ФЗ в России для того, чтобы лекарство попало на рынок, необходимо пройти процедуру государственной регистрации. В ходе нее заявитель оформляет и направляет в контролирующее ведомство множество документов о своем продукте и многократно доказывает, что он отвечает актуальным нормативам по качеству, безопасности и действенности. Однако до недавнего времени производители должны были проходить еще и процедуру сертификации, которая фактически ориентирована на те же цели. А дополнительные контролирующие меры – это, конечно, трата финансовых ресурсов и времени, в течение которого препарат мог бы уже лежать на прилавках и приносить прибыль производителю.
Форма обязательной сертификации лекарственных средств
Процедура обязательной сертификации лекарственных средств в России выполняется в форме регистрации медицинских препаратов. Она проводится Росздравнадзором по инициативе заявителя, в качестве которого выступают как сами производители таких продуктов, так и организации, занимающиеся ее распространением. При этом регистрация становится обязательной для большей части лекарств, созданных для широкого обращения на рынке фармацевтических товаров.
Процедура государственной регистрации
Сейчас законодатели пришли к выводу, что тех проверочных мер, которые входят в процедуру госрегистрации, будет достаточно для организации адекватного контроля за этим сегментом рынка. Она по-прежнему будет выполняться с соблюдением регламента, прописанного в приказе Минздрава от 21 сентября 2016 года N 725н. В роли государственного ведомства, которое оказывает эту услугу, выступает Росздравнадзор.
Сама процедура регистрации включает два базовых этапа:
- подготовительный. На этом шаге всю работу делает заявитель. Правда, он может обратиться в надзорное ведомство за консультацией, если она ему потребуется – например, для организации клинических исследований. В течение подготовительной фазы заявитель проходит проверку Минпромторга на соответствие правилам надлежащей производственной практики, составляет регистрационное досье в формате общетехнического документа (ОТД) и проводит клинические исследования препарата. Выполнение каждой процедуры должно быть подтверждено документально. Сформированный пакет документов представляется в Росздравнадзор для инициирования процедуры госрегистрации;
- основной. На этом шаге в действие вступают две основные государственные структуры. В роли координатора процесса функционирует Росздравнадзор ,который проверяет документы, полученные от заявителя, и составляет задание на выполнение необходимых экспертиз. Сами проверки и исследования в рамках этих экспертиз проводит отдельная уполномоченная организация – ФГБУ «НЦЭСМП». Она работает только по заданию Росздравнадзора, то есть прямые контакты с заявителями для нее запрещены. В рамках этого этапа проводится два типа экспертиз: проверка безопасности и качества препарата, а также проверка по шкале «риск-польза». В процессе этого исследования выполняется сопоставление предполагаемого риска, обусловленного использованием препарата, и ожидаемой пользы от его применения в конкретной врачебной ситуации.
Получение регистрационного удостоверения
Изделие, которое прошло все требуемые проверки и подтвердило свое соответствие актуальным нормативам по качеству и безопасности этой категории товаров, получает регистрационное удостоверения. Оно становится документальным подтверждением того, что конкретное лекарство зарегистрировано и имеет прав продаваться на рынке. Информация об этом также отражается в едином государственном реестре. Он опубликован в открытом доступе на официальном портале Министерства здравоохранения.
Хранение сертификатов
Отраслевой
стандарт «правила отпуска ЛС в аптечных
организациях» утвержденный приказом
80 содержит два постулата подтверждающих
качество товара
- Не
подлежат приемке ЛС и другие товары, и
товары с истекшим сроком годности, не
соответствующе требованиям качества
и без документов удостоверяющих их
качество - По
требованию покупателя заведующий
аптекой предоставляет информацию
документов по ценам и срокам годности
лс и других товаров, а также и о документах
подтверждающих качество ЛС.
Поставка
ЛС и товаров в аптеку осуществляется с
использованием сопроводительного
пакета документов: договор поставки
между поставщиком и аптекой, счет( счет
идет на оплату в бугалтерию за
осуществленную поставку ЛС в аптеку,
счет фактура (в бугалтерию аптеки для
оплаты за поставлены товар), товарно
-транспортная накладная или товарная
накладная-)-на основании этого документа
осуществляется приемка товара в аптеки
по количеству и качеству, договор
поставки заключается в двух экземплярах(
один экземпляр поставшику второй
покупателю), счет и счет фактуа в одном
экземпляре в аптеку поставляется а
товарная накладная- в трех или четырех
экземплярах(один поставщику, второй
покупателю, третий – в случае поставки
НС И ПВ для учета), протокол согласования
оптовых цен на жизненно необходимые
ЛС- в двух экземплярах( один поставщику,
оптовику а второй покупателю-аптеке)
Документы
качества: копии сертификата
соответствия, сан и пид заключение,
регистрационное удостоверение или
реестр сертификатов. В реестре сертификата
указывается наименование и сериям НС,
срок годности, номер сертификата и дата
его выдачи и срок действия сертификата.
Сертификаты
соответствия и декларация действуют
до истечения срока годности на ЛС и
хранятся в аптеке(нормативных документах
подтверждающих сроки хранения сертификатов
нет, но сертификаты соответствия хранятся
пять лет.) документы качества продукции
являются неотъемлемой частью
сопроводительного пакета документов
и должны хранится в бугалтерии-5 лет. Но
поскольку нормативного акта утверждающего
сроки хранения данных документов нет,
сертификаты, удостоверения регистрационные
и сан и пид заключения хранятся до сих
пор, пока товар не продан. Хранят
сертификат соответствия в аптеках двумя
способами. По дате прихода товара и
алфавиту. При использовании любого
способа хранения документов найти в
нужный момент документ не составит
труда, на ценнике товара всегда есть
дата его поступления в продажу, данная
информация имеется в компе.
Если
аптека использует системы электронного
документа оборота, то все сертификаты
хранятся в единой компьютерной базе,
единственный минус такой организации
работы- отсутствие на распечатке
сертификата печати поставщика и подписи
ответственного лица. В аптеке за хранение
сертификатов ответственность возлагается
на руководителей. ЛС без сертификата
соответствия не подлежат реализации
на территории РФ. При поставках в аптеки
лс должны сопровождаться сертификатами
или декларации соответствия или их
копиями, заверенными держателем
поддлеников сертификата соответствия
или декларации, нотариусом, органом по
сертификации выдавшим сертификат, либо
указанием в товарно сопроводительных
документах (товарная накладная), номера
сертификата срока его действия, и органа
его выдавшего, эти документы заверяются
синей печатью поставщика или производителя
и подписью с указанием его адреса и
телефона. На такие товары аптечного
ассортимента- перевязочный материал,
атомарные биноы, резины, стекла, фарфора;
а также товары соприкасающиеся с телом
человека, дез средства, лечебная
косметика, медицинская техникагоь-
требуется наличие сертификата
соответствия, выдаваемого ростестстандартом.
На отдельные товары: БАД, пищевые, и ЛС,
дрожи, мин.вода, продукты детского
питания, продукты лечебного и
профилактического питания, лечебные
косметические средства, в том числе
детского ассортимента, средства гигиены
полости рта, медицинская техника,
гигиенические средства, требуется сан
и пид заключение.
Декларации
о соответствии ЛС в праве принимать
зарегистрированные юридические и
физические лица, являющиеся изготовителями
или продавцами, на основании договора
с ними. Декларация о соответствии ЛС
принимается декларантом на основании
собственных доказательств, и доказательств
полученных с участием третьей стороны
(центр сертификации), в качестве
доказательств полученных с участием
третьей стороны являются протоколы
испытаний проведенные в акредитованном
испытательном центре и имеющие сертификаты
соответствия на производство или систему
качества системы сертификации гос
россии(два последних документа должны
быть у производителя) в этом случае
производитель может принимать декларацию
о соответствии самостоятельно.
Хранение
декларации или ее заверенной копии
осуществляется у декларанта не менее
трех лет после окончании срока ее
действия.
Наотропы:
пирацетам, ноотропил, принимается 0,8
три раза в день
Фенотропил
0,1 три раза в день 2-3 месяцев
В соответствии
с приказом №428 от 08.11.2006 года «об
обеспечение качества лекарственных
средств территории челябинской области».
В соответствии с данным приказом все
лекарственные средства фальсифицированные
контрофактные, регистрируются в журнале
«баланс движения забракованных или
фальсифицированных лекарственных
средств в организациях оптовой торговли
и аптечных организациях».
Данный
журнал ведётся руководителем аптеки в
случае поступления писем от поставщиков,
которые поставили не доброкачественные
лекарственные средства. Не качественные
лекарственные средства хранятся в
карантинной зоне специально выделенной
для этой цели (это обычно секция шкафа
промаркированная «карантинная зона»).
Не доброкачественные поставленные ЛС
возвращаются поставщиком. Заведующая
аптекой ежедневно отслеживает информацию
о фальсифицированных ЛС на сайте в
интернете www/regmet.ru.
фальсифицированные ЛС являются угрозой
национальной безопасности.
Проблема
обращения фальсифицированных ЛС на
фарм рынке России достигло таких
масштабов что представляет реальную
угрозу национальной безопасности.
Наибольшее
количество фальсифицированных ЛС
реализуется через интернет аптеки.
От того на
сколько работники аптечных организаций
добросовестно и ответственно выполняют
свои функции и обязанности не малой
степени зависит предотвращение их
попадания к покупателю.
Ранее
государство являлось единоличным
участником всей системы обращения ЛС.
Начинает с процесса разработки,
производства, и всех последующих этапов
их доведения до потребителя. В настоящее
время по сведениям роз здрав надзор в
стране действует более 600 предприятий
по производству ЛС различных форм
собственностей, которые удовлетворяют
соответствующие потребности внутреннего
рынка. Отсутствие достаточного объема
и ассортимента ЛС отечественного
производства компенсируется продукцией
возимой из за рубежа. До 1998 года в России
проблемы фальсификатов не существовало.
Первый
официально зарегистрированный случай
фальсификат в России относится к 1997
году, когда был выделен поддельный
кровезаменитель полиглюкин, производство
завода красфарм.
В 2000 году
в России впервые были официально
подтверждены минздравом факты попадания
в розничную сеть фальсифицированных
ЛС- нистатин, ношпа, сумманет.
По оценкам
экспертов доля фальсификата на Российском
фарм рынке в настоящее время достигает
от 10-15%. Каждая 10 упаковка. По оценкам
минздрава за 2012 год который приводится
татьяной голиковой количество
фальсификатов составляет 0,4%.
Не легальный
бизнес не обошел своим вниманием фарм
рынок, особенно в части изготовления и
реализации контрафактной продукции.
Перестройка экономики и внутреннего
рынка в 90-х годах затронула в первую
очередь фарм рынок России.
Фальсификат-
это подделка, поддельный продукт не
имеющий сертификата. На мировом фарм
рынке при общем его объеме 300 миллиардов
долларов доля подделок составляет около
14-20 миллиарда долларов. В странах Африки
и латинской Америки доля продающихся
фальсифицированных лекарств достигает
30-50%.
Сертификация лс.
Цель
сертификации-
защита прав интересов потребителей,
проведение единой государственной
политики в области обеспечении населения
высококачественными ЛС.
Сертификация
состоит из 2 частей:
-сертификация
лекарства, его соответствие требования
стандарта.
Сертификат
соответствия ЛС-
документ, удостоверяющий безопасность
и соответствие качества ЛС.
Сертификат
выдают центры контроля качества,
аккредитованные и имеющие лицензию на
данный вид деятельности.
Лекарственные
средства подлежат обязательной
сертификации. Перечень продукции, в
отношении которой законодательными
актами Российской Федерации предусмотрена
обязательная сертификация, установлен
Постановлением Госстандарта Российской
Федерации от 30 июля 2002 года №64 «Номенклатура
продукции, в отношении которой
законодательными актами Российской
Федерации предусмотрена обязательная
сертификация». В данный Перечень включены
медикаменты, химико-фармацевтическая
продукция и продукция медицинского
назначения.
Постановлением
Госстандарта Российской Федерации от
24 мая 2002 года №36 утверждены Правила
проведения сертификации в системе
сертификации лекарственных средств
системы сертификации ГОСТ (далее Правила
№36).
«Обязательной
сертификации подлежат лекарственные
средства:
выпускаемые
предприятиями-производителями
лекарственных средств на территории
Российской Федерации;
ввозимые
на территорию Российской Федерации в
порядке, установленном действующим
законодательством».
Группы
лекарственных средств, не подлежащие
обязательной сертификации, приведены
в Письме Государственного комитета
Российской Федерации по стандартизации
и метрологии от 15 января 2003 года
№ИК-110-25/110 «О лекарственных средствах,
не подлежащих сертификации»:
«В
порядке информации сообщаю, что в
соответствии с областью применения
«Правил проведения сертификации в
Системе сертификации лекарственных
средств Системы сертификации ГОСТ »,
утвержденных Постановлением Госстандарта
России от 24 мая 2002 года №36, и Законом
Российской Федерации «О защите прав
потребителей» не подлежат обязательной
сертификации следующие группы
лекарственных средств:
лекарственные
средства без индивидуальной упаковки
, не предназначенные для розничной
продажи;
фармацевтические
субстанции для производства лекарственных
средств;
иммунобиологические
препараты, вакцины, сыворотки (не вошедшие
в список товаров, для которых требуется
подтверждение проведения обязательной
сертификации)».
Сертификат
соответствия лекарственного средства
оформляется органами по сертификации
лекарственных средств после проверки
лекарственного средства на соответствие
требованиям нормативных документов,
утвержденных федеральным органом
исполнительной власти в сфере
здравоохранения, на заявителя.
Сертификат
качества лекарственного средства –
документ, подтверждающий соответствие
качества лекарственного средства
государственному стандарту качества
лекарственных средств (статья 4 Закона
№86-ФЗ);
Срок
действия сертификата на партию (серию)
лекарственных средств не устанавливается.
Сертификат действителен при поставке,
продаже партии продукции в течение
срока годности лекарственного средства,
установленного нормативными документами.
Порядок
проведения сертификации лекарственных
средств включает в себя:
представление
заявки в орган по сертификации;
рассмотрение
заявки и представленных заявителем
документов;
принятие
решения по заявке, выбор схемы сертификации;
сертификацию
систем качества (производства), если
это предусмотрено схемой сертификации;
анализ
полученных результатов испытаний,
проверок и принятие решения о выдаче
(об отказе в выдаче) сертификата
соответствия;
оформление
и выдачу сертификата соответствия;
осуществление
инспекционного контроля за сертифицированной
продукцией (если это предусмотрено
схемой сертификации);
корректирующие
мероприятия при нарушении соответствия
продукции установленным требованиям
и неправильного применения знака
соответствия;
Сертификация
лекарственных средств процедура не
бесплатная, в соответствии с пунктом 4
статьи 23 Федерального закона от 27 декабря
2002 года №184-ФЗ «О техническом регулировании»
(далее Закон №184-ФЗ) работы по обязательной
сертификации подлежат оплате заявителем.
Участниками
системы сертификации являются:
-орган
управления системой сертификации ЛС
при МЗ
-центральный
орган по сертификации. В него входят:
институт государственного контроля
ЛС и институт стандартизации.
-центры
контроля качества на территории РФ
Документальная
часть по сертификации:
-нормативно-организационные
и нормативно-методические документы к
которым относятся положение о системе
сертификации ЛС, приказы и инструкции.
-ГФ,
международная и зарубежные фармакопеи
Значение экспертизы лекарств
Выполнение экспертизы для регистрации лекарственных средств – это ключевое условие для обеспечения легитимности оборота медицинских препаратов в России. Именно по ее результатам уполномоченный орган принимает решение об удовлетворении заявления о регистрации либо отказе в выдаче регистрационного удостоверения. При этом порядок обращения за предоставлением государственной услуги по регистрации лекарственных средств (РЛС) устанавливается положениями приказа Минздрава от 21 сентября 2016 года N 725н. Координацию взаимодействия участников процедуры и принятие окончательных решений по этому вопросу согласно приказу осуществляет Министерство здравоохранения. Однако в цепочке процедур, составляющих алгоритм госрегистрации, заняты и другие органы.
Обзор документа
С 29 ноября 2019 г. действуют законодательные поправки по вопросу ввода в гражданский оборот лекарственных препаратов для медицинского применения.
Из сферы действия Закона о техническом регулировании исключены разработка, принятие, применение и исполнение требований по обращению лекарственных средств.
Кроме того, лекарственные препараты исключены из перечней сертифицируемой и декларируемой продукции.